Stages¶
The workflow/ directory contains everything necessary to run the Drug Sniffer
pipeline conveniently on a variety of computing platforms.
Container configuration can be found in the containers/ directory. However,
Drug Sniffer users should not need to interact with the contents of this
directory.
The individual components of the pipeline (stages) are described below. Although it is possible to use the pipeline without understanding the individual stages, this information is provided here to aid users in troubleshooting, and for completeness.
Docker Images¶
Each stage in the Drug Sniffer pipeline has its own Docker image. The images can
be built all at once with the build-images.sh script found in the tool/
directory, which can be run from the repository root. This script accepts two
parameters as environment variables, listed below. The default values should be
acceptable for most users.
IMAGE_NAMESPACE- the owner of the image, this is usually a project or organization name and, again, doesn’t matter for images that will never be pushed to a registryIMAGE_VERSION- the version identifier to be applied to the image
To build the image for a particular stage for testing purposes, run
build-image.sh in the directory corresponding to that stage. To run the
container, for manual debugging, use run-image.sh. To run the automated
smoke tests, use test-image.sh. The tests rely on data files stored in the
test/ directory and may write additional outputs to that same location
when they run.
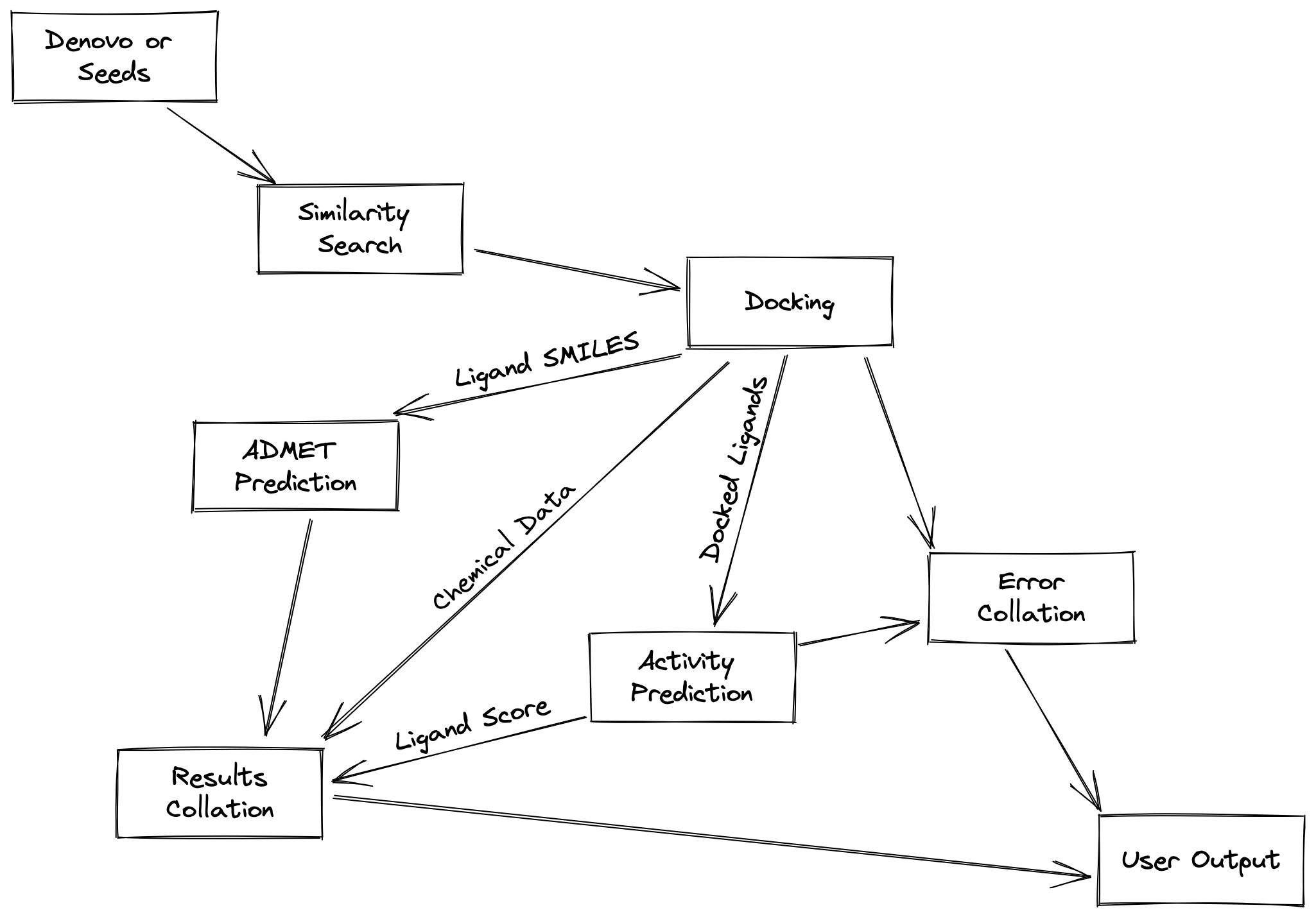
The diagram above illustrates how data flows through the Drug Sniffer pipeline. For the most part, users will not need to concern themselves with this.
Running a Stage¶
Within each Docker image is a script, included in the PATH, called
run.sh. This script serves as the API for the container. To run a stage, the
user sets certain environment variables and then executes the script. All stages
work this way. Required and optional environment variables are described along
with each stage below.
Stage 1 - Target Identification¶
The user is presumed to have chosen a target protein. For example, the SARS-CoV-2 Nucleocapsid Protein.
Stage 2 - Pocket Prediction¶
Pocket prediction is a manual process. Once a pocket has been selected, it is provided to the Drug Sniffer pipeline as a PDB file along with information about the pocket geometry. See Parameters for details.
Some common pocket prediction tools include ProBis, FTMAP, and POCASA.
Stage 3 - Denovo Molecule Design¶
One option for identifying leads is to dock a virtual library of pre-enumerated compounds. Alternatively, one may evolve drug-like molecules using a genetic algorithm.
In Drug Sniffer, we make use of AutoGrow4, an open-source program that uses an evolutionary algorithm to generate novel leads from a set of chemically diverse molecular fragments. The de novo process makes use of in silico chemical reactions to generate new compounds, and the population of compounds is iteratively refined over a number of cycles.
In order to remove compounds with undesirable physical and chemical properties, AutoGrow4 makes use of molecular filters such as PAINS and Lipinski rules. The molecules from the last three generations are set up as seeds to identify similar molecules in massive libraries.
Required environment variables:
RECEPTOR_PATH- path to the original PDB file containing the proteinpocket chosen manually by the user
CENTER_X- the x-coordinate center of the pocketCENTER_Y- the y-coordinate of the pocketCENTER_Z- the z-coordinate of the pocketSIZE_X- the size of the pocket in the x directionSIZE_Y- the size of the pocket in the y directionSIZE_Z- the size of the pocket in the z direction
Optional environment variables:
SOURCE_COMPOUND_FILE- the list of compounds used to seed Autogrow4 (default is Fragment_MW_up_to_250.smi, a custom version of the files found in Autogrow4)DOCKING_EXHAUSTIVENESS- Autogrow4 exhaustiveness (default is 1)NUMBER_OF_PROCESSORS- the number of CPUs to use for Autogrow4 (default is 4)NUMBER_OF_GENERATIONS- the number of generations to use with Autogrow4 (default is 10)
Dependencies (included in Docker image):
Autogrow4
Version: 4.0.3
Autodock Vina
Website: https://vina.scripps.edu
Version: 1.1.2
The Docker container for this stage also uses Miniconda 4.10.3 running Python 3.8 to supply a Python interpreter and various dependencies for Autogrow4, which is in line with the recommended way to use this software.
Stage 4 - Similarity Search¶
In order to expand the candidate pool, the denovo molecules are used as seeds to
identify other similar structures in larger databases. After building 1024-bit
ECFP4 fingerprints for the denovo molecules, they are compared against molecules
in the database. The fingerprints for the database molecules are pre-generated
and are referenced with the molecule_db parameter. The result of this
stage is a collection of molecules likely to be similar to the denovo molecules
and therefore (hopefully) likely to fit the receptor.
Required environment variables:
SEED_LIGANDS_SMI- a .smi file containing the ligands created in stage 3MOLECULE_DB- the path to a molecule database to match denovo ligands against; the database format is described alongside the other parameters
Optional environment variables:
TANIMOTO_CUTOFF- the minimum Tanimoto score for a match between a denovo ligand and a molecule in the database, molecules above this score will be sent on to stage 5 (default is 0.5)OUTPUT_PATH- the directory to which molecule .smi files should be written (default is ./output)
This stage consists entirely of custom code but relies on RDKit (specifically the Python bindings), version 2021.9.4.
The output is one or more .smi files that contain the SMILES strings,
taken from the molecule database, thought to match the binding pocket.
Each line contains one SMILES string, then the database it came from, the
name of the molecule within that database, and the offset it was stored at
in the database (which is only for debugging purposes). The fields for each
molecule are separated by tab characters.
Stage 5 - Protein Ligand Docking¶
For the seed-neighbor molecules identified by the similarity search, optimized structures (lowest energy conformation generated using OpenBabel) of neighbors are docked into their respective targets using AutoDock Vina. The number of docking poses produced and the exhaustiveness parameter for the search for each ligand are parameterized by the user; the default values are 9 and 4, respectively.
The Autodock Vina seed value is set to 42 in order to facilitate reproduction.
This stage is allowed to fail since OpenBabel sometimes fails to produce a 3D structure. These failures are ignored because there is no reasonable way to recover and the consequences are generally insignificant.
Required environment variables:
RECEPTOR_PDBQT- path to the receptor (pocket) chosen by the user, in PDBQT formatCENTER_X- the x-coordinate center of the receptorCENTER_Y- the y-coordinate of the receptorCENTER_Z- the z-coordinate of the receptorSIZE_X- the size of the receptor in the x directionSIZE_Y- the size of the receptor in the y directionSIZE_Z- the size of the receptor in the z directionLIGANDS_SMI- a file containing the ligands (molecules) chosen for further processing in stage 4
Optional environment variables:
NUMBER_OF_POSES- the maximum number of docking poses to attempt using Autodock Vina
Dependencies (included in Docker image):
Autodock Vina
Website: https://vina.scripps.edu
Version: 1.1.2
Stage 6 - Activity Prediction¶
The docking score produced by AutoDock Vina is only a loose estimate of the actual binding affinity. DrugSniffer adds 3 post hoc re-scoring methods (1) the Autodock Vina score (2) the SMINA score (3) dock2bind (the default) which is a neural network re-scoring strategy. The model is trained on ligand-protein complexes taken from the LIT-PCBA and DUD-E.
For each docked pose, 16 pose descriptors calculated by SMINA, along with the DFIRE estimate of protein–ligand potential are used as input to the model. dock2bind produces a value from 0 to 1 and can be thought of as the model’s confidence that the molecule binds to the pocket, constrained by the specific pose.
The model accepts the values below, in order, as a comma- or whitespace-delimited table:
Pose (identifier)
gauss_1
gauss_2
repulsion
hydrophobic
non_hydrophobic
vdw
non_dir_hbond_lj
non_dir_anti_h_bond_quadratic
non_dir_h_bond
acceptor_acceptor_quadratic
donor_donor_quadratic
electrostatic
ad4_solvation
ligand_length
constant_term
num_tors_div
DFIRE
The output of this stage is a comma-delimited table of values containing the columns listed below:
Pose (identifier)
Model output (from 0 to 1)
Required environment variables:
RECEPTOR_PATH- path to the original PDB file containing the protein receptor (pocket) chosen manually by the userDOCKED_PDBQT- the docked ligand as a PDBQT file that contains all poses computed by Autodock Vina in stage 5
Dependencies (included in Docker image):
Autodock Vina
Website: <https://vina.scripps.edu>
Version: 1.1.2
DLIGAND2
Commit: 03b0347d450b1a70f4728d1d170626100b585bb4
Smina
Open Babel
Website: http://openbabel.org/wiki/Main_Page
Installed from Debian repositories
Stage 7 - ADMET Prediction (optional)¶
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) of drugs plays a key role in determining which among the potential candidate structures are to be prioritized. The ADMET prediction here is based on molecular fingerprint-based predictive models. While a majority of the models are binary classification models, for some endpoints such the metabolic intrinsic clearance, acute oral toxicity in rats, plasma protein binding and elimination half-life, multiclass models are proposed.
For a complete list of the models employed see https://doi.org/10.1186/s13321-021-00557-5. For classification models, two additional values are reported: a confidence (how certain the model is that the prediction is a singleton) and a credibility. A confidence value of 0.95 suggests that the classifier is quite certain that the prediction is likely to be a single label. A relatively low value of credibility suggests that the compounds are not sufficiently represented in the training set and that the user needs to treat the prediction with caution.
Required environment variables:
LIGAND_SMI- path to the ligand under evaluation
Optional environment variables:
ADMET_CHECKS- space-separated list of ADMET checks for FPADMET, values in the range[1, 56](default is empty)
Dependencies (included in Docker image):
FPADMET
Commit: d61d63e3d3c37e887a5d4b1959260d9f1b41f77a
Stage 8 - Error Collation¶
Errors that occur in certain stages (those that tend to produce recoverable errors) are assembled into a single report and written to the path provided by the output_dir parameter.
Stage 9 - Results Collation¶
Results are assembled into a single file and written to the path provided by the output_dir parameter.
