Drug Sniffer¶
Drug Sniffer is a virtual screening (VS) pipeline capable of screening billions of molecules using only thousands of CPU hours, using a novel combination of ligand-based (LBVS) and structure-based (SBVS) methods.
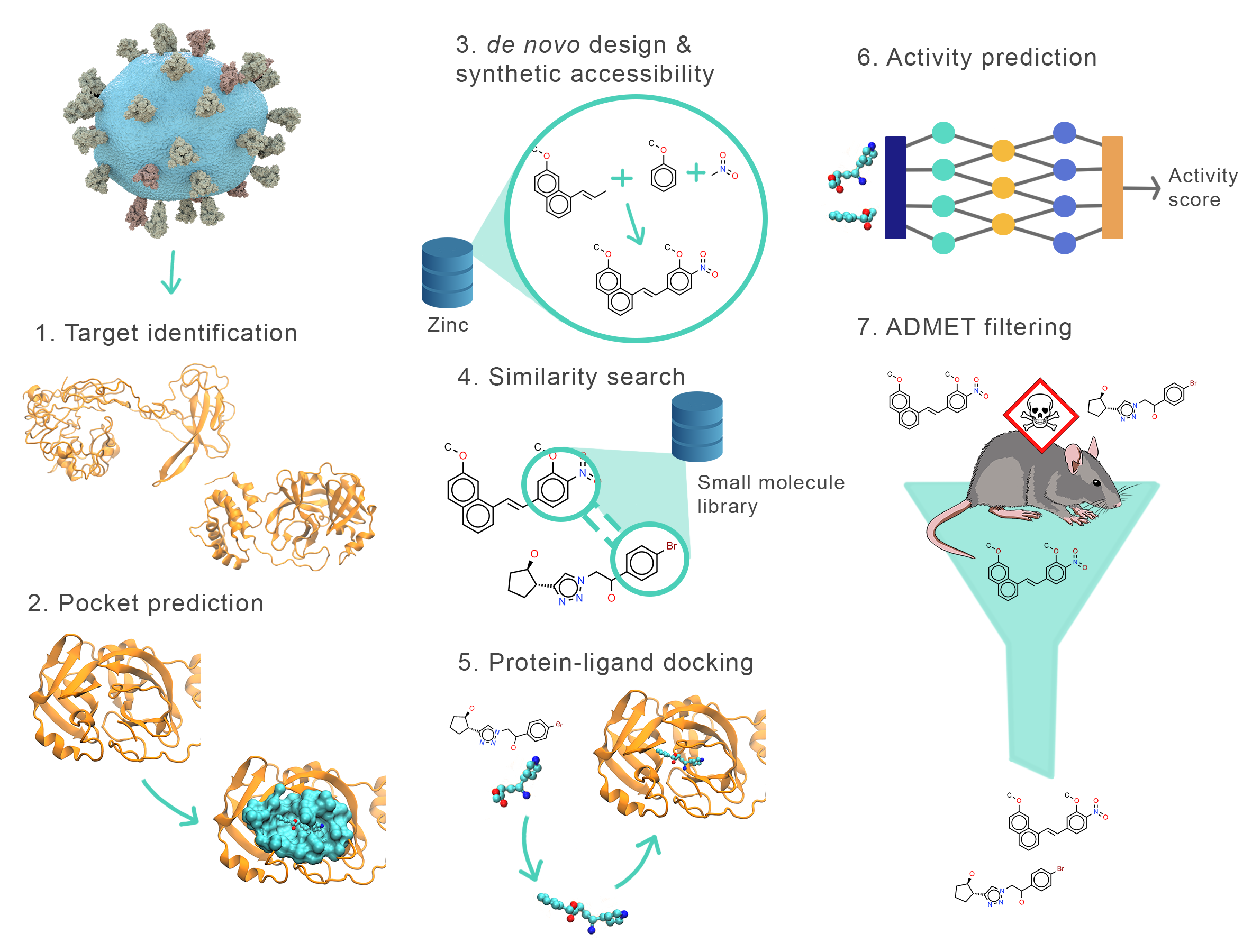
The pipeline requires the user to identify the structure and pocket of the target protein (stages 1 and 2). These stages are completed manually by the user.
Then, the pipeline begins by designing multiple de novo ligands for the identified binding pockets (stage 3). Next, it uses these ligands as seeds to identify similar compounds from a small-molecule database (stage 4). The resulting neighbors are then subjected to rigid-body docking (stage 5) and re-ranked with a new scoring model (stage 6).
Optionally, the pipeline also allows the user to run possible ligands through FP-ADMET, an ADMET filter (stage 7).
The Drug Sniffer pipeline has been implemented as a Nextflow workflow. Each stage has a corresponding script and Docker image that are used to execute the computations contained in the stage.
See the Usage guide for details on how to run Drug Sniffer.
Code¶
Code can be found on GitHub. Feel free to create an “issue” on GitHub if you find a bug or need support.
Cite¶
If you use drugsniffer, please cite the paper.
Venkatraman, T. H. Colligan, G. T. Lesica, D. R. Olson, J. Gaiser, C. J. Copeland, T. J. Wheeler, and A. Roy, “Drugsniffer: An open source workflow for virtually screening billions of molecules for binding affinity to protein targets,” Front. Pharmacol., vol. 13, Apr. 2022.
Table of Contents¶
- Usage
- Database
- Stages
- Docker Images
- Running a Stage
- Stage 1 - Target Identification
- Stage 2 - Pocket Prediction
- Stage 3 - Denovo Molecule Design
- Stage 4 - Similarity Search
- Stage 5 - Protein Ligand Docking
- Stage 6 - Activity Prediction
- Stage 7 - ADMET Prediction (optional)
- Stage 8 - Error Collation
- Stage 9 - Results Collation
- Parameters
- Errors
- Troubleshooting
Indices¶
Acknowledgements¶
This research was supported in part by:
Research Council of Norway (Grant No. 262152)
National Institutes of Health (NIH), Department of Health and Human Services under BCBB Support Services Contract HHSN316201300006W/HHSN27200002 to MSC, Inc.
NIH grant R01GM132600
DOE grant DE-SC0021216.
The Drug Sniffer software was developed under a collaboration between Amit Roy, Vishwesh Venkatraman, and members of the Wheeler Lab ,
